易基因:ChIP-seq等揭示植物H3K27me3相关转座元件沉默模式及与DNA甲基化动态转换:Genome Biol
大家好,这里是专注表观组学十余年,领跑多组学科研服务的易基因。
转座元件(transposable elements,TEs)是基因组中能够移动和复制的重复序列,其活动通常对宿主生物有害,因此被宿主严格调控。然而,TEs的活动在进化中也扮演了重要角色,例如通过为附近基因提供遗传或表观遗传调控模块来影响转录程序。TEs沉默通常与高DNA甲基化相关,而在植物中,TEs的甲基化可以发生在CG、CHG和CHH三种序列环境中,并与组蛋白H3K9二甲基化(H3K9me2)耦合,这两种表观遗传标记共同抑制TEs的表达和转座,从而保证基因组完整性。另一方面,H3K27me3与基因转录抑制相关,通常与发育、生殖或代谢和应激反应相关的蛋白编码基因有关。尽管H3K27me3和DNA甲基化通常被认为是互斥的系统,分别特异性靶向基因和TEs的转录沉默,但有研究表明在某些情况下,这两种标记可以共存并协同作用。
近日,法国巴黎萨克雷大学细胞综合生物学研究所Angélique Déléris团队以模式生物拟南芥为研究对象,研究了植物中转座元件(TEs)的表观遗传沉默机制,特别是与H3K27me3(组蛋白H3K27三甲基化)相关的沉默状态。研究结果揭示了在野生型拟南芥中,许多TEs可以被H3K27me3单独靶向并沉默(而不依赖DNA甲基化沉默机制)。此外,研究还发现了TEs在不同拟南芥自然生态型之间存在表观遗传状态的转换,即从DNA甲基化到H3K27me3的相互转换,这一现象被称为“bifrons”。相关研究成果以“️Alternative silencing states of transposable elements in Arabidopsis associated with H3K27me3”为题发表于《️Genome Biology》(IF:10.1)期刊。

标题:Alternative silencing states of transposable elements in Arabidopsis associated with H3K27me3(拟南芥中与 H3K27me3 相关的转座因子可变沉默状态)
发表时间:2025-01-20
期刊:Genome Biology
影响因子:IF10.1/Q1
展开全文技术平台:ChIP-seq、BS(易基因金牌技术)
️研究摘要
DNA/H3K9甲基化和Polycomb蛋白组(Polycomb-group,PcG)-H3K27me3沉默通路长期以来被认为在哺乳动物、植物和真菌中是互斥的,并且分别特异性地作用于转座元件(TEs)和基因。但即使在DNA/H3K9甲基化机制缺失情况下,许多TEs仍然可以被H3K27me3靶向,有时甚至与DNA甲基化共存。
本研究结果揭示了在野生型拟南芥植物中,TEs也可以被H3K27me3单独靶向并沉默。这些被H3K27me3标记的TEs不仅包括退化残迹,还包括看似完整的拷贝,它们显示出响应性PcG靶基因的表观遗传特征以及活跃的H3K27me3调控。同时,H3K27me3可以以TE特异性方式沉积在新插入的转基因TE序列上,表明沉默由顺式( cis)作用决定。最后通过比较拟南芥的自然生态型发现了一类TEs(称为“bifrons”),它们的标记取决于生态型,要么被DNA甲基化标记,要么被H3K27me3标记。这种差异可以追溯到TEs的内在特征以及反式作用因子,并揭示TEs在其生命周期中表观遗传状态变化。
总之,本研究揭示了开花植物中与H3K27me3相关的可变TE沉默模式,同时还表明了在物种水平上两种表观遗传标记(DNA甲基化和H3K27me3)之间存在动态转换,这一新的范式可能扩展到其他多细胞真核生物。
️研究方法
(1)ChIP-seq技术:检测H3K27me3在TEs上的分布情况。
(2)BS技术:用于分析TEs的DNA甲基化水平。
️结果图形
️(1)在野生型拟南芥植物中,许多TE只由H3K27me3标记
研究发现,拟南芥基因组中有超过4000个TEs被H3K27me3标记,这些TEs不仅包括退化的残迹,还包括看似完整的拷贝。这些TEs显示出与响应环境或发育信号的经典PcG靶基因相似的染色质特征,其H3K27me3模式受到组蛋白H3K27去甲基化酶的积极调控。
根据H3K27me3和DNA甲基化的覆盖长度,将TEs分为四类。其中,“class 3”仅被H3K27me3标记,占TE总数的15%,但仅占TE碱基的8.4%。这些TEs倾向于分布在短TE类别中(尤其是小于1kb的TE),而长TEs(大于3.5kb)通常与DNA甲基化相关,尽管一小部分长TEs(大于3.5kb)仅被H3K27me3标记。
这些TEs在基因组中的分布与DNA甲基化的TEs不同,它们不仅存在于异染色质区域,还分散在常染色质臂和近着丝粒区域。
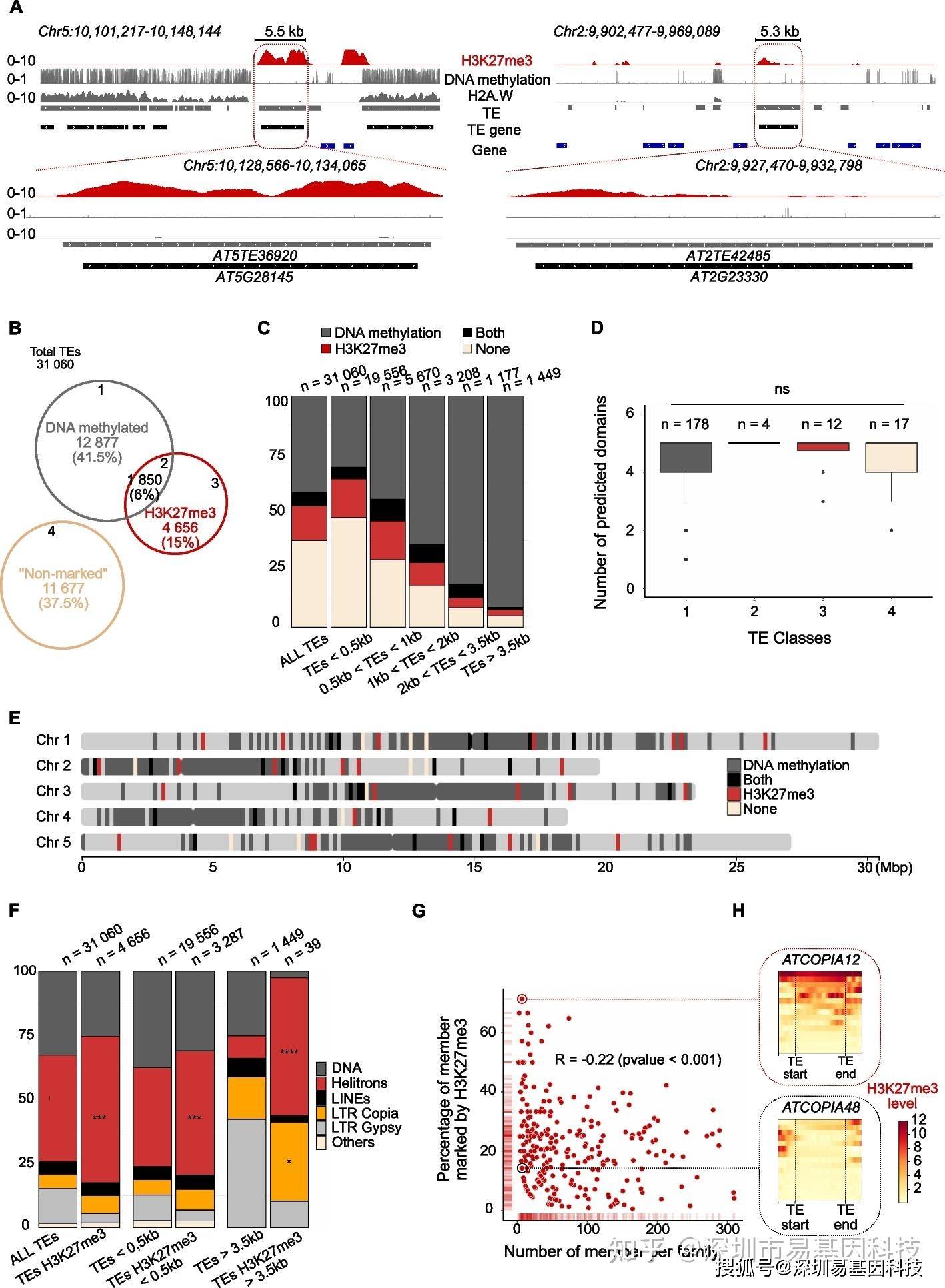
图1:在野生型拟南芥(Col-0)中,许多转座元件(TEs)被H3K27me3标记。
- 代表性基因组浏览器视图显示野生型Col-0中两个被H3K27me3标记的TEs的H3K27me3(红色)、DNA甲基化(灰色)和组成型异染色质组蛋白变体H2A.W(灰色)水平。基于表观遗传修饰的TE分类的维恩图。当CG甲基化在TE长度上的平均值>50%时,TE被定义为“DNA甲基化”(类别1-2)。当H3K27me3 peaks至少覆盖TE长度的20%时,TE被定义为处于H3K27me3状态。按TE长度分类的TE类别的分布堆叠条形图。对大于3.5 kb的TE进行LTR Copia预测结构域数量箱线图,ns=不显著。显示大于3.5 kb的TE在基因组中分布示意图。按TE长度分类的所有TEs或H3K27me3标记的TEs的TE超家族分布堆叠条形图。显示TE亚家族成员被H3K27me3标记的百分比与TE拷贝数量之间的相关性图。基于每个TE拷贝的H3K27me3水平的热图。上图:ATCOPIA12;下图:ATCOPIA48。
️(2)H3K27me3标记的TEs具有沉默且响应的基因表观遗传特征,其转录受PcG抑制
H3K27me3标记的TEs中有23%和22%分别含有H2A.Z或H2AK121ub标记,这些标记通常与动态转录抑制基因相关。这表明这些TEs具有沉默但可响应的基因特征。
在arp6(H2A.Z沉积缺失)和atbmi(H2AK121ub沉积缺失)突变体中,部分TEs的H3K27me3水平降低,表明H3K27me3的沉积依赖于H2A.Z和H2AK121ub。此外,在ref6 elf6 jmj13三重突变体中,许多H3K27me3标记的TEs显示出更高的H3K27me3水平,表明这些TEs的H3K27me3模式受组蛋白去甲基化酶调控。
在swn clf PRC2双突变体中,约2/3的H3K27me3标记的TEs显著上调,表明H3K27me3在拟南芥中可以转录抑制TEs表达。
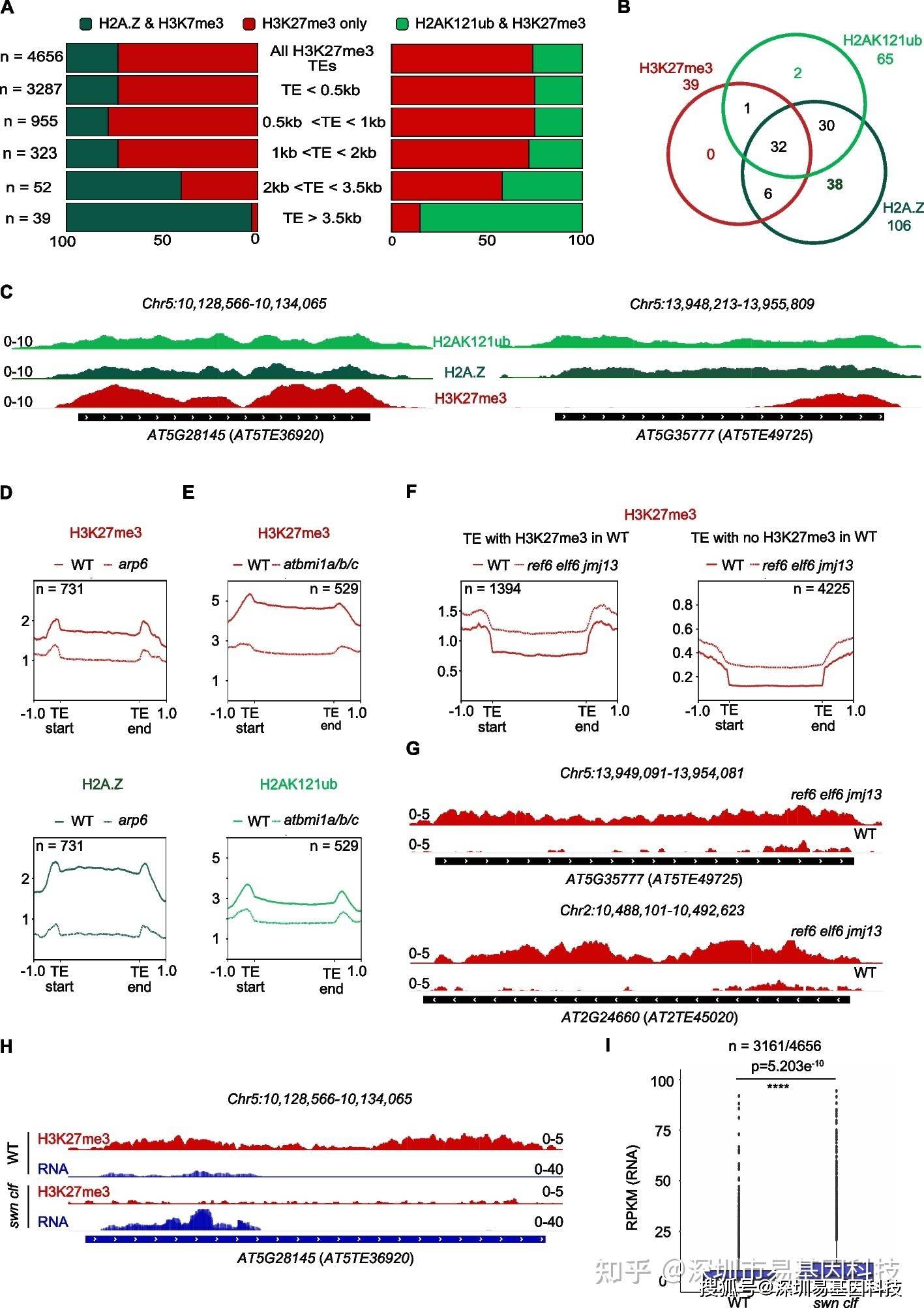
图2:H3K27me3在TEs上可与H2AUb和H2A.Z相关联,表现出活跃的去甲基化和转录抑制。
- 堆叠条形图显示在H3K27me3标记的TEs中,按大小范围分类的被H2A.Z变体或H2AK121泛素化标记TEs的分布情况。如果一个TE同时存在H3K27me3和H2A.Z或H3K27me3和H2AK121ub的峰重叠,则该TE被认为共标记。维恩图显示在大于3.5 kb的TEs上H3K27me3、H2A.Z变体和H2AK121ub的存在情况。代表性基因组浏览器视图展示来自第3类(被H3K27me3标记)的2个TEs的ChIP-seq数据,显示了野生型(WT)中的H2AK121ub(浅绿色)、H2A.Z(深绿色)和H3K27me3(红色)标记。Metagenes图显示了在WT和arp6突变体(该突变体在H2A.Z掺入受影响的TE亚集中存在缺陷)中H3K27me3(上图)和H2A.Z(下图)的水平。Metagenes图显示了在WT和atbmi1a/b/c突变体(该突变体在H2AK121ub沉积受影响的TE亚集中存在缺陷)中H3K27me3(上图)和H2AK121ub(下图)的水平。Metagenes图显示了在WT和ref6 elf6 jmj13三重H3K27me3去甲基化酶突变体中H3K27me3的水平。左侧图表示在WT中已被H3K27me3标记但在ref6 elf6 jmj13中该标记增加的TEs。右侧图表示在WT中未被H3K27me3标记但在ref6 elf6 jmj13中该标记显现的TEs。代表性基因组浏览器视图展示了来自第3类的2个TEs在野生型和ref6 elf6 jmj13中的H3K27me3(红色)标记。代表性浏览器视图展示TE在野生型和swn clf双突变体中的H3K27me3(红色)和RNA(蓝色)。箱线图显示与野生型相比,在swn clf双突变体中显著上调的第3类TEs(4656个中有3161个)。
️(3)H3K27me3标记以顺式决定方式被招募到新插入的转基因TEs上
通过将三个不同的COPIA LTR逆转录转座子序列克隆到二元载体中,并将其插入植物基因组,研究新插入的TEs是否能够招募H3K27me3。结果表明,这些新插入的TEs能够以序列特异性的方式招募H3K27me3,表明H3K27me3的招募依赖于TE序列本身的内在特征。
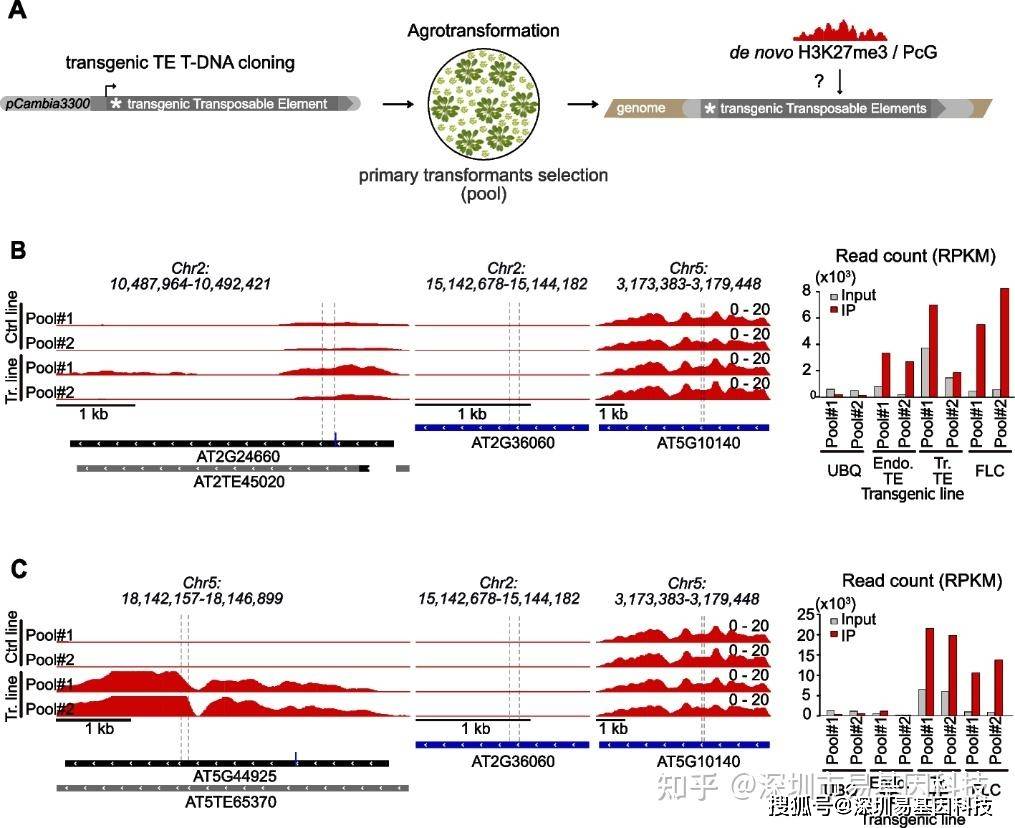
图3:新插入的TE序列可以以一种指导性的方式从头招募H3K27me3。
- 实验方案:将单个TE序列克隆到二元载体中,将其引入植物基因组,并在T1代的大规模植株群体(15-20株)中通过H3K27me3 ChIP-seq进行研究,以稀释位置效应。左侧图:代表性基因组浏览器视图显示在两个独立的植物群体中,H3K27me3的平均分布(IP-INPUT)在COPIA12B序列(顶部图)上,这些植物要么被转化了无关的转基因(Ctrl),要么被转化了感兴趣的TE。在“Ctrl”轨迹中的H3K27me3信号反映内源TE的H3K27me3状态。在“Tr.”轨迹中的H3K27me3信号反映内源+转基因的H3K27me3状态:与对照线相比更高的信号表明除了内源基因外,转基因TE上也招募了H3K27me3。转基因群体之间的变异性反映了转基因TE拷贝数的差异。中间图:UBQ和FLC分别作为H3K27me3招募的阴性和阳性对照。右侧图:通过在引入SNP的区域对内源和转基因COPIA12B序列进行H3K27me3免疫沉淀的定量来确定转基因上的招募。左、中和右图与B中的COPIA21序列相同。
️(4)群体水平上的TE中在DNA甲基化和Polycomb标记之间转换
通过比较拟南芥不同自然生态型(如Col-0和KBS-Mac-74)的H3K27me3和DNA甲基化数据,发现存在一类TEs(称为“bifrons”),其在不同生态型中表现出表观遗传状态的转换,即从DNA甲基化到H3K27me3或反之。
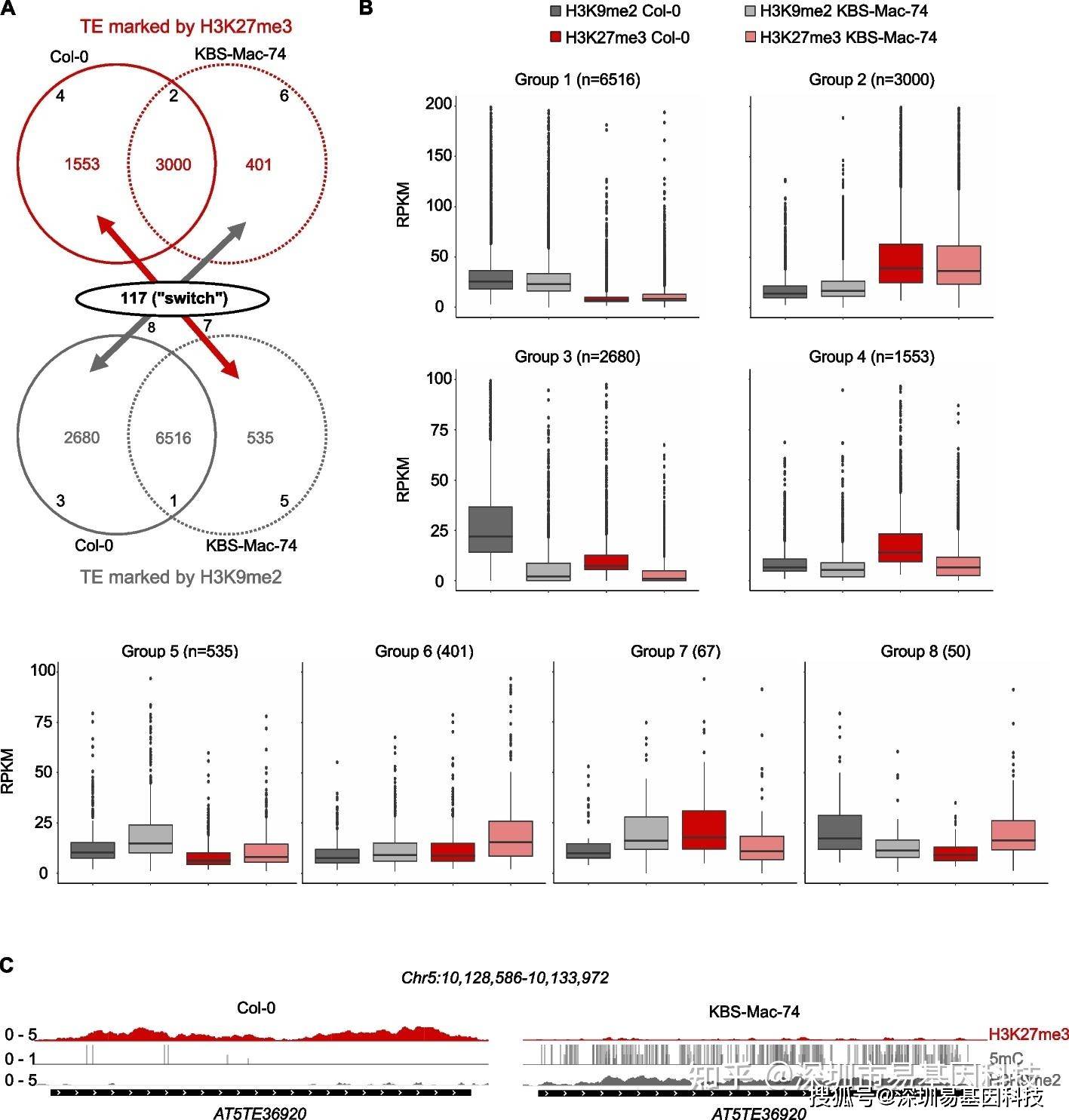
图4:通过不同生态型之间的比较,可视化TEs上沉默表观遗传标记之间的转换。
- 维恩图显示在两种不同生态型(Col-0和KBS-Mac-74)中,被H3K27me3标记的TEs(顶部,红色数字)和被DNA甲基化标记的TEs(底部,灰色数字)。两个维恩图的交集显示了在一种生态型中被H3K27me3标记而在另一种生态型中被H3K9me2标记的TEs(107个)。盒须图显示在Col-0和KBS-Mac-74中,八个不同TE簇的H3K9me2水平(以RPKM表示)和H3K27me3水平。代表性基因组浏览器视图显示在Col-0和KBS-Mac-74两个生态型中,一个同源TE拷贝的H3K27me3和DNA甲基化水平(红色:H3K27me3,深灰色:CG甲基化,灰色:CHG甲基化,浅灰色:CHH甲基化,黑色条:TE注释)。
️(5)某些TEs上H3K27me3的沉积与TE的顺式决定因子以及反式作用因子在遗传上相关
通过对近500个拟南芥自然生态型的DNA甲基化数据进行全基因组关联研究(GWAS),发现TEs的表观遗传状态转换与TE序列本身(cis-determinants)以及参与DNA甲基化的转录因子(trans factors)相关。
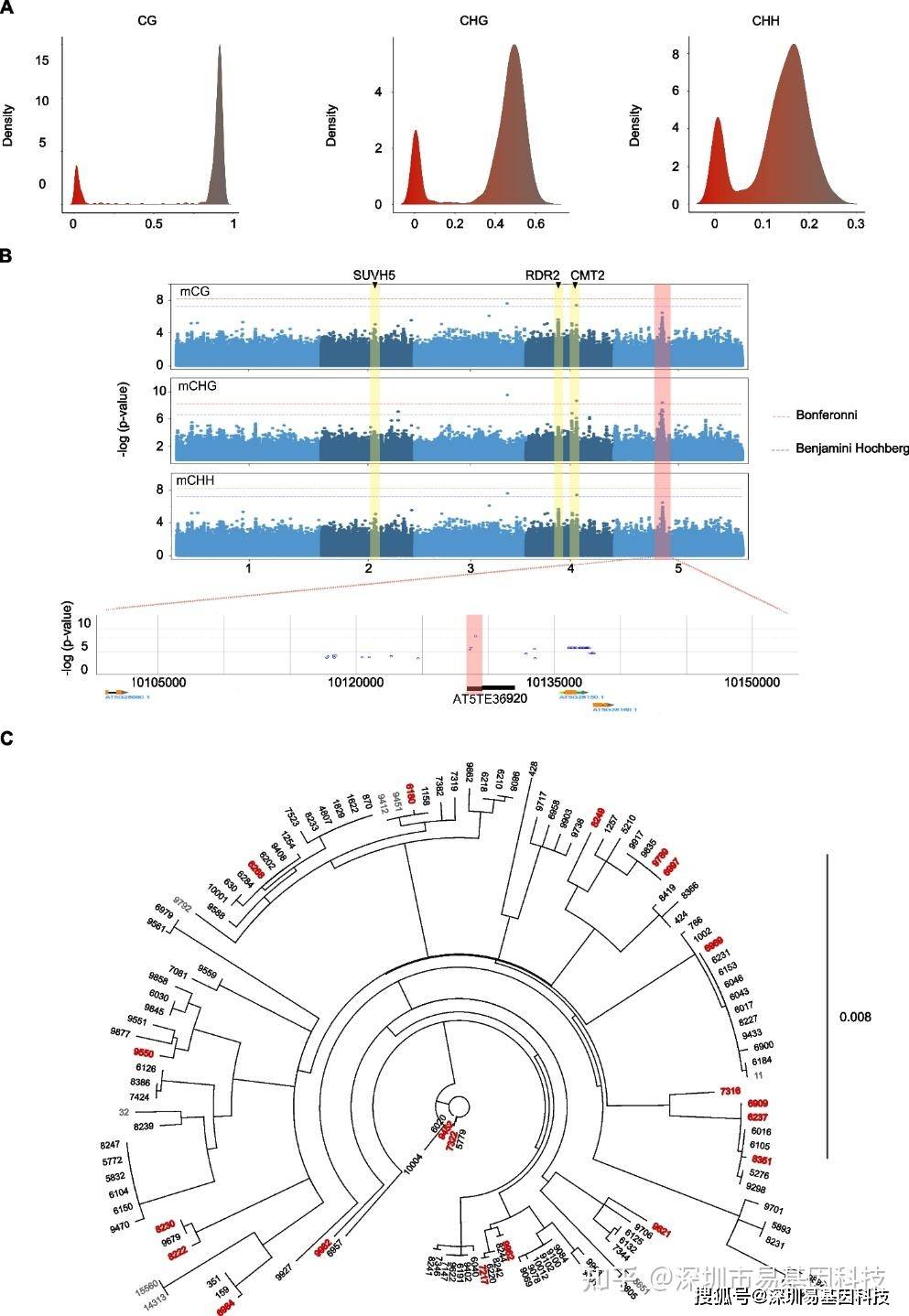
图5:表观遗传转换的遗传决定因子。
- 图表显示了一个同源TE,即COPIA12D(AT5TE36920)的DNA甲基化值,以及在三个序列环境中(CG、CHG和CHH)的甲基化密度分布情况。曼哈顿图展示了靶向COPIA12D(AT5TE36920)的单变量全基因组关联研究(GWAS)结果,分别为mCG(上图)、mCHG(中图)和mCHH(下图)。水平线表示全基因组显著性水平。黄色框表示与DNA甲基化相关的反式作用因子的峰值,粉色框表示TE的顺式区域。130个不同生态型中AT5TE36920序列的系统发育树。右侧标示了遗传距离。红色加粗的生态型表示COPIA12D无DNA甲基化,被认为被H3K27me3标记。黑色的生态型表示COPIA12D存在DNA甲基化。灰色的生态型表示没有关于COPIA12D表观遗传状态的信息。
️易小结
本研究揭示了通过ChIP-seq等技术拟南芥中TEs的一种与H3K27me3相关的可变沉默机制。此外,研究还发现了TEs在物种水平上表观遗传状态的动态转换,这一现象可能扩展到其他多细胞真核生物。这些发现挑战了传统观念,即拟南芥TEs仅通过DNA甲基化进行沉默,并为理解TEs在进化中的作用提供了新的视角。
ChIP-seq和BS测序技术在本研究中的作用
ChIP-seq技术:用于检测H3K27me3在TEs上的分布情况,揭示了TEs的表观遗传特征以及H3K27me3与H2A.Z和H2Aub的共定位。通过ChIP-seq,研究人员鉴定出哪些TEs被H3K27me3标记,并分析这些标记的动态变化。
BS技术:用于分析TEs的DNA甲基化状态,与ChIP-seq数据相结合,帮助研究人员理解TEs的表观遗传状态转换。通过亚硫酸盐测序,研究人员能够确定TEs在不同生态型中DNA甲基化差异,从而揭示表观遗传状态转换的分子基础。
️关于易基因染色质免疫共沉淀测序 (ChIP-seq)
染色质免疫共沉淀(Chromatin Immunoprecipitation,ChIP),是研究体内蛋白质与DNA相互作用的经典方法。将ChIP与高通量测序技术相结合的ChIP-Seq技术,可在全基因组范围对特定蛋白的DNA结合位点进行高效而准确的筛选与鉴定,为研究的深入开展打下基础。
DNA与蛋白质的相互作用与基因的转录、染色质的空间构型和构象密切相关。运用组蛋白特定修饰的特异性抗体或DNA结合蛋白或转录因子特异性抗体富集与其结合的DNA片段,并进行纯化和文库构建,然后进行高通量测序,通过将获得的数据与参考基因组精确比对,研究人员可获得全基因组范围内某种修饰类型的特定组蛋白或转录因子与基因组DNA序列之间的关系,也可对多个样品进行差异比较。
应用方向:
ChIP 用来在空间上和时间上不同蛋白沿基因或基因组定位
- 转录因子和辅因子结合作用复制因子和 DNA 修复蛋白组蛋白修饰和变异组蛋白
技术优势:
- 物种范围广:细胞、动物组织、植物组织、细菌微生物多物种富集经验;微量建库:只需5ng以上免疫沉淀后的DNA,即可展开测序分析;方案灵活:根据不同的项目需求,选择不同的组蛋白修饰特异性抗体。
技术路线:

️关于植物DNA甲基化研究
️(1)植物中的DNA甲基化
对于植物而言,面对生长环境的改变,表观遗传的变异会改变植物DNA的构象,从而改变染色质和蛋白质的结构,达到调节基因组的作用。研究发现,当植物面临生物胁迫和非生物胁迫时,植物基因组中DNA甲基化会发生改变,并且这些改变会遗传给后代。所以DNA甲基化的改变能够丰富植物物种的多样性,加强植物的环境适应性。
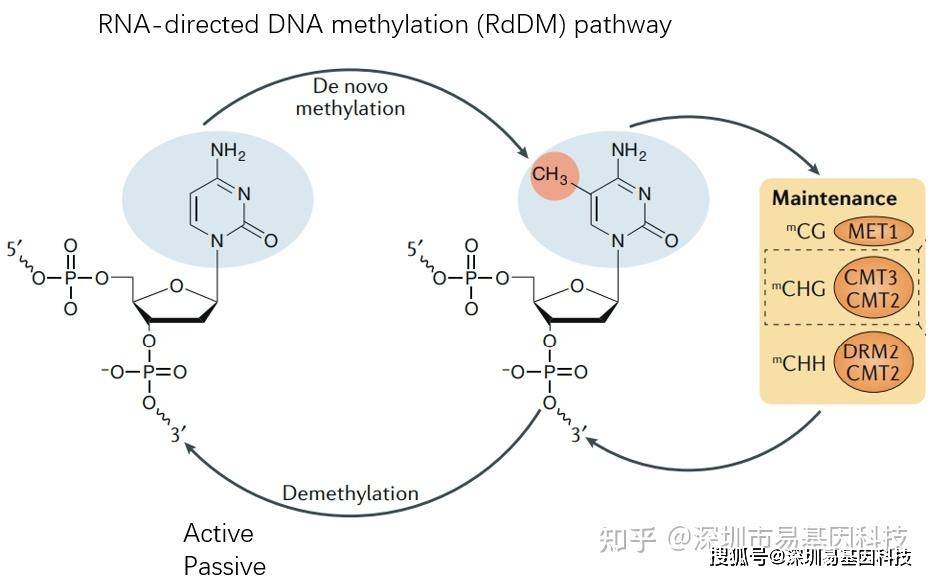
不同于哺乳动物基因组只有CG甲基化,植物基因组甲基化有CG,CHG,CHH(H代表任何非G的碱基)甲基化。而维持这三种不同的DNA甲基化的分子机制非常复杂。
️(2)植物中的DNA甲基化研究技术

️(3)植物DNA甲基化研究思路
植物DNA甲基化一般遵循三个步骤进行数据挖掘。首先,进行整体全基因组甲基化变化的分析,包括平均甲基化水平变化、甲基化水平分布变化、降维分析、聚类分析、相关性分析等。其次,进行甲基化差异水平分析,筛选具体差异基因,包括DMC/DMR/DMG鉴定、DMC/DMR在基因组元件上的分布、DMC/DMR的TF结合分析、时序甲基化数据的分析策略、DMG的功能分析等。最后,将甲基化组学&转录组学关联分析,包括Meta genes整体关联、DMG-DEG对应关联、网络关联等。
易基因提供全面的表观基因组学(DNA甲基化、DNA羟甲基化、cfDNA)和表观转录组学(m6A、m5C、m1A、m7G、ac4C、RNA与蛋白互作)、DNA与蛋白互作及染色质开放性技术方案(ChIP-seq、ATAC-seq),详询易基因:0755-28317900。

️参考文献:
Hure, V., Piron-Prunier, F., Yehouessi, T. et al. Alternative silencing states of transposable elements in Arabidopsis associated with H3K27me3. Genome Biol 26, 11 (2025). https://doi.org/10.1186/s13059-024-03466-6


